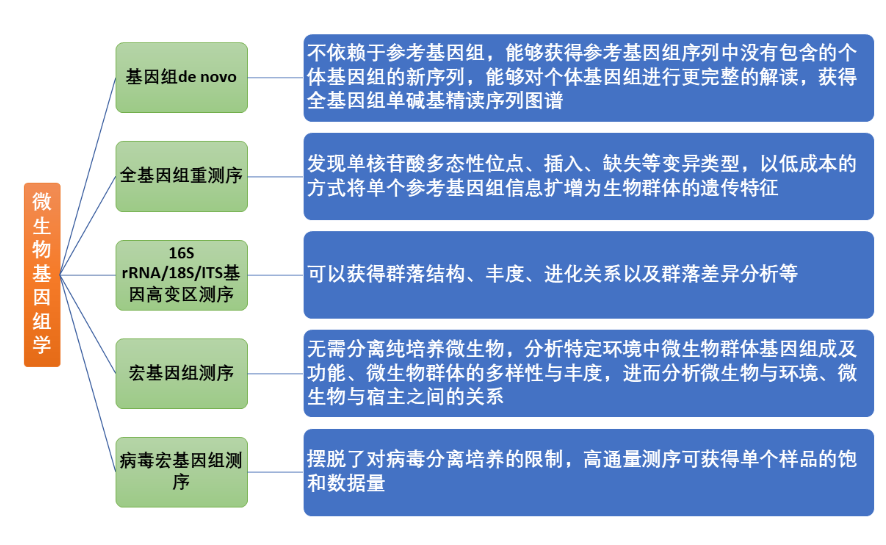
生物信息(Bioinformatics)摘要
- 简介:是信息科学领域和生命科学领域的交叉学科,是以计算机为主要工具,以大量生物数据库和分析软件为基础的学科,也是为解决生物学问题为目标,为人类揭示生命奥秘提供一条新的途径的学科。
- 生物范式的转变:In vivo→In vitro→In silico
- 生物信息学学科发展的黄金时期:Human genome project(HGP)
- 基本方法:数据库检索、建立数据库
- 研究内容:收集、整理、储存、加工、发布和分析生物学数据;发展新的数理和信息科学的技术和方法用于管理和分析生物数据。
生物信息学数据库
- 关于Database(数据库):收集、整理、储存、加工、发布和检索数据的系统。(序列、结构、生物分子互作、基因表达、文献著作等)
- 核苷酸数据库
- 定义:DNA、mRNA、tRNA、rRNA序列(RNA序列以cDNA形式收集),氨基酸序列(CDS编码序列)
- 三大核苷酸数据库:GenBank(NCBI—美国)、ENA(欧洲)、DDBJ(日本)(www.insdc.org)
- NCBI: http://www.ncbi.nlm.nih.gov/
- GenBank:建立和管理分子生物学和生物医学领域的公用数据库,包括部分蛋白质序列,源于核酸序列注释结果(https://www.ncbi.nlm.nih.gov/genbank/)
-
序列格式:GenBank Flat File格式,每条序列有三个专有的编号或者标识(identifier)—Locus name(大多以登录号代替)、Accession number(注册号或登录号)、GI等
- EST:收录5‘端或3’端的cDNA序列;cosmid/BAC/YAC外源插入片段的末端序列;转座子插入位点的侧翼序列、Alu PCR序列(https://www.ncbi.nlm.nih.gov/nuccore)
- TSA:非实验直接获得,由源自不同克隆的多条序列经生物信息学软件拼接得到的(http://www.ncbi.nlm.nih.gov/genbank/TSA.html)
- TPA:可以是一条已有序列的一部分或者由多条序列经生物信息学软件拼接得到(http://www.ncbi.nlm.nih.gov/genbank/tpa)
- HTGS:尚未完成测序的重叠群(>2kb)的序列(https://www.ncbi.nlm.nih.gov/genbank/htgs/)
- Genome基因组数据库:测序完成和正在测序物种基因组序列(RefSeq)、遗传图、物理图、序列组装、基因注释等(https://www.ncbi.nlm.nih.gov/genome)
- 单核苷酸多态性数据库:利于发现致病基因、进化分析等(https://www.ncbi.nlm.nih.gov/snp)
- 基因组结构变异数据库:基因组大片段的插入、缺失和拷贝数的变异(https://www.ncbi.nlm.nih.gov/dbvar)
- Gene数据库https://www.ncbi.nlm.nih.gov/gene
- RefSeq数据库:https://www.ncbi.nlm.nih.gov/refseq/

- EMBL-EBI: http://www.ebi.ac.uk/
- DDBJ: http://www.ddbj.nig.ac.jp/
- NCBI: http://www.ncbi.nlm.nih.gov/
- 国家基因组科学数据中心(NGDC): http://bigd.big.ac.cn
- 启动子数据库:包括部分cis-element信息,以及人类、小鼠、果蝇、斑马鱼、线虫等大量启动子信息(https://epd.vital-it.ch/index.php)
- miRNA数据库:收集了大量不同物种的hairpin precursor miRNA序列;可通过miRNA名称、关键词、染色体位置等来检索;可分析一条DNA序列中是否可能包含miRNA(http://www.mirbase.org/)
- **蛋白质数据库
- UniProt:来源于实验的有详细注释的序列(SwissProt)和自动注释序列(TrEMBL) ,可用关键词(Text search)和序列比对(BLAST similarity search)进行检索(http://www.uniprot.org/)
- UniRef100:非冗余的UniProt蛋白质序列
- UniRef90:聚类UniRef100中一致性超过90%且80%重叠的蛋白质,取最长的一条(序列数压缩58%)
- UniRef50:聚类UniRef90中一致性超过50%且80%重叠的蛋白质,取最长的一条(序列数压缩79%)
- UniProtKB:(注意:查询修饰看PTM,查询可被什么酶剪切也是PTM,跨膜结构域是subcellular,Mutagenesis是Pathology)在数据库主页选择框选择UniProtKB库,使用关键词检索——可根据物质、关键词、功能、通路进行筛选;可检索蛋白质相关结果
- UniRef:提供序列簇、以及序列的同源异构体的信息(序列比对后相似性高的聚集在一起)
- UniParc:所有的序列压缩成一个作为代表
- Proteome:蛋白组,研究所有蛋白的学科
- PIR:信息整合的蛋白质序列数据(https://pir.georgetown.edu)
- PRO:给一个蛋白定义,便于了解其与另一个蛋白的关系(如作用、代谢相关的元素)
- iPTMnet:系统了解蛋白在翻译后有什么修饰
- iProLINK:检索蛋白关键的注释信息(iProClass)、分类信息(PIRSF)

- PRF:可检索已发表在杂志上的蛋白质序列的修饰位点、S-S键等(http://www.prf.or.jp/)
- PDBSTR:可检索蛋白序列和二级结构、α碳结构(http://www.genome.jp/dbget-bin/www_bfind?pdbstr)
- Prosite:可检索蛋白质的蛋白质家族以及结构域(https://prosite.expasy.org/)
- PKR:可对已知蛋白激酶的序列进行比较、分类;检索蛋白质激酶的三维结构以及与疾病相关的蛋白激酶(http://www.kinase.com/)
- 结构数据库
- PDB(Protein Data Bank):可通过关键词(或者注册号)或BLAST系统检索蛋白质结构,有多种检索方式、多种结构显示视图、多种结构/序列分析
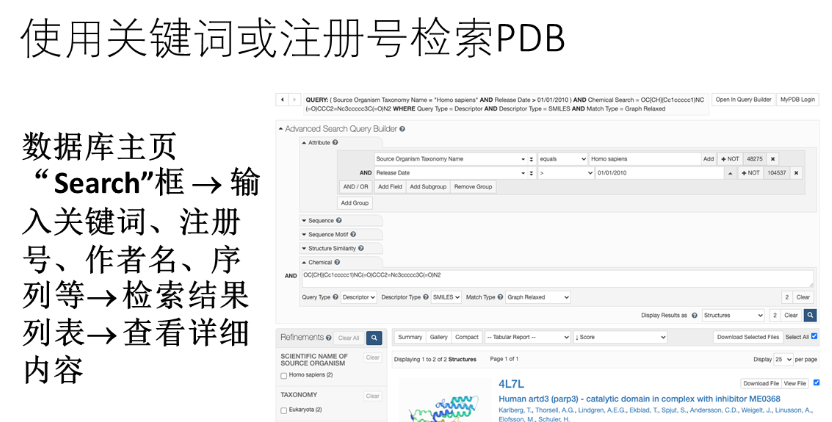
- NDB(Nucleic Acid Database):可检索DNA或RNA的三级结构
- PDIdb(Protein-DNA Interface Database):DNA-蛋白质复合体的X射线衍射结构及分类(http://melolab.org/pdidb/web/content/home)
- PDB(Protein Data Bank):可通过关键词(或者注册号)或BLAST系统检索蛋白质结构,有多种检索方式、多种结构显示视图、多种结构/序列分析
- 酶代谢数据库
- KEGG:各种代谢、遗传等路径图;可检索参与各种路径的基因(https://www.kegg.jp)
- KEGG PATHWAY:各种代谢、遗传等路径图,可检索参与各种路径的基因

- KEGG BRITE:

- KEGG MODULE:模块,关键词检索
- KEGG REACTION:生化反应
- KEGG PATHWAY:各种代谢、遗传等路径图,可检索参与各种路径的基因
- MetaCyc(Metabolic Pathway Database):代谢通路(https://metacyc.org/)
- Plant Metabolic Pathway Database:https://www.plantcyc.org/
- KEGG:各种代谢、遗传等路径图;可检索参与各种路径的基因(https://www.kegg.jp)
- 物种分类数据库:
- Taxonomy数据库:(https://www.ncbi.nlm.nih.gov/taxonomy )

- Taxonomy数据库:(https://www.ncbi.nlm.nih.gov/taxonomy )
- 文献数据库
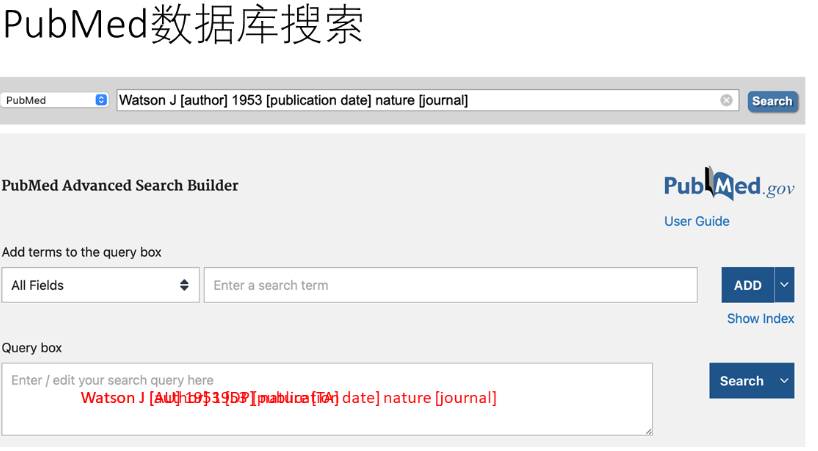
- PubMed:美国国家医学图书馆的数据库——收录医学、分子生物学、基础生物学
- OMIM:NCBI的数据库,收录人类基因、遗传疾病相关文献(https://www.ncbi.nlm.nih.gov/omim)
- Agricola:美国农业部农业图书馆的数据库,农业刊物(https://agricola.nal.usda.gov/) 注意:常用的序列格式为:FASAT、Genbank、EMBL、数据库后台存储格式ASN.1、其他格式
- 格式转换:Sequence Format Conversion(https://www.ebi.ac.uk/Tools/sfc/)
生物信息学数据库检索
- 检索数据库的方法:
- 关键词或词组为基础的数据库检索
- 名词、描述性词、词组
- 序列注册号(Accession number)
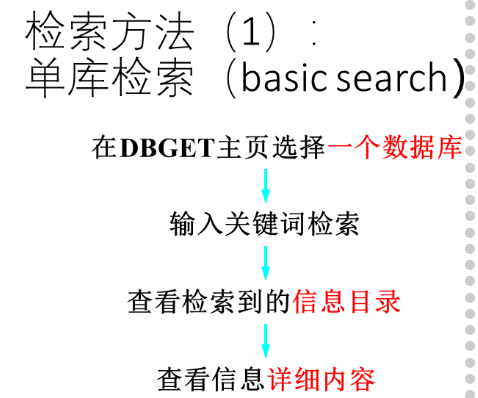
- 检索体系:NCBI Entrez、EBI search、Integrated database retrieval system(DBGET)
-
检索须知1:

-
检索须知2:
 例:核苷酸数据库中有多少条检索注册号在AF123456-AF123478之间?—用冒号来表示范围,记得用中括号表示accession。
例:核苷酸数据库中有多少条检索注册号在AF123456-AF123478之间?—用冒号来表示范围,记得用中括号表示accession。- NCBI的检索体系:可对6大类35个数据库进行检索(https://www.ncbi.nlm.nih.gov/search/)![[Pasted image 20220528213547.png]]
 除上述两方法,也可通过Advanced Search进行数据检索。
除上述两方法,也可通过Advanced Search进行数据检索。 - PubMed:

- NCBI的检索体系:可对6大类35个数据库进行检索(https://www.ncbi.nlm.nih.gov/search/)![[Pasted image 20220528213547.png]]
- EBI-search:检索面宽,但操作复杂—18大类148个数据库与该体系相连(http://www.ebi.ac.uk/)![[Pasted image 20220528215008.png]]

- DBGET(Integrated database retrieval system): 与KEGG相连,操作较简单,但检索面较EBI search 简单(http://www.genome.jp/dbget/

- 核苷酸和蛋白质序列为基础的数据库检索—通过序列对位排列,突出相似的结构区域的方法。(如两条序列比对时,不相似的部分(空位插入或缺失)会用虚线表示)
- 序列比对用途:基因预测;分析基因或蛋白质的功能;分析物种演化;检测突变;插入或缺失;序列延长;序列定位;基因表达谱分析
- 对位排列分析的种类:
- 序列对库对位排列分析:从数据库(核苷酸数据库、蛋白质数据库)中寻找同源序列
- 两序列对位排列分析
- 多序列对位排列分析
- 序列对位排列分析的基本原理
- 记分矩阵:长度一定时,分数越高,两条序列匹配的越好


- 空位(间隔)罚分:序列对位排列分析时允许插入空位

- 记分矩阵:长度一定时,分数越高,两条序列匹配的越好
- 对位排列的方法:点阵分析、动态规划、词或K串方法
- 关键词或词组为基础的数据库检索
- 序列对库对位排列分析:
- 基本概念
- 关于序列相似性

- Sequence identity(相同):两条序列在同一位点上的核苷酸氨基酸残基完全相同
- Sequence similarity(相似):两条序列在同一位点上的氨基酸残基的化学性质相似(相似性是positive)
- Homology(同源):当你有超过100个氨基酸的序列或超过100个核苷酸的序列时,同源性的概念分别为25%的氨基酸序列相同,70%的核苷酸序列相同。
- 关于alignment的global与local:

- 关于alignment的gapped或者ungapped:
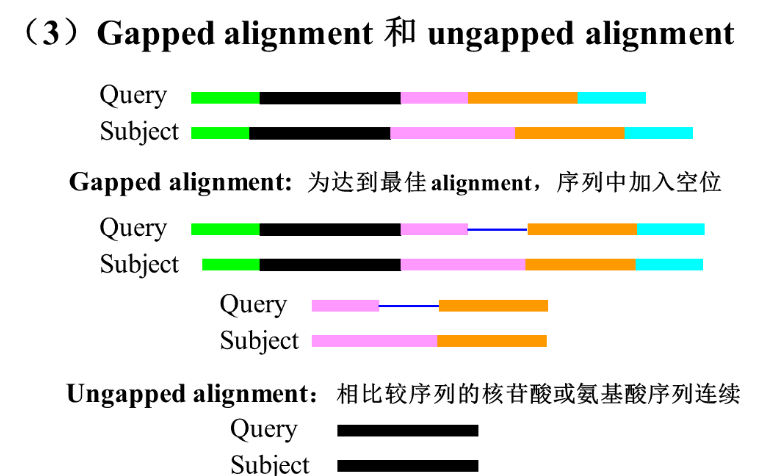
- Alignment score和E(expect)values:
 关于E-value:
关于E-value:
- Low-complexity regions(LCRs):

- 关于序列相似性
- 检索体系:
- BLAST(Basic Local Alignment Search Tool):https://blast.ncbi.nlm.nih.gov/Blast.cgi
- blastn:用核苷酸序列检索核苷酸数据库

- blastp:用蛋白质序列检索蛋白质数据库

- blastx:将核苷酸序列通过6种阅读框翻译成不同的蛋白质序列检索蛋白质数据库

- tblastn:用蛋白质序列检索核苷酸数据库(数据库中的序列被翻译出不同的蛋白质序列)
- tblastx:将核苷酸序列通过6种阅读框翻译成不同的蛋白质序列检索核苷酸数据库(数据库中的序列被翻译出不同的蛋白质序列)
- PSI-BLAST(Position Specific Iterated BLAST):
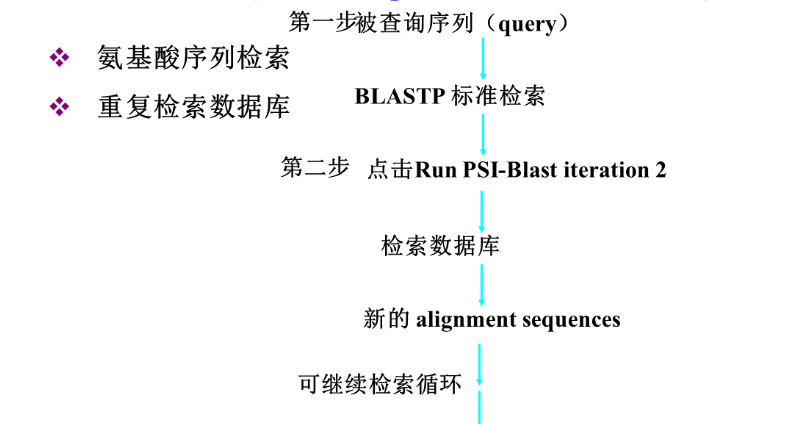
- PHI-BLAST(Pattern Hit Initiated):

- CD-Search:检索蛋白质保守结构域的数据库(https://www.ncbi.nlm.nih.gov/cdd/)![[Pasted image 20220529151434.png]]
- Primer-BLAST:设计PCR引物,分析引物特异性((https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome))
- Multiple Alignment:多重比对,进化分析((https://www.ncbi.nlm.nih.gov/tools/cobalt/cobalt.cgi?LINK_LOC=BlastHomeLink))
- 本地BLAST:

- blastn:用核苷酸序列检索核苷酸数据库
- FASTA检索:(http://www.ebi.ac.uk/Tools/sss/)
- 两序列对位排列分析:(http://blast.ncbi.nlm.nih.gov/)![[Pasted image 20220529153053.png]]结果格式:
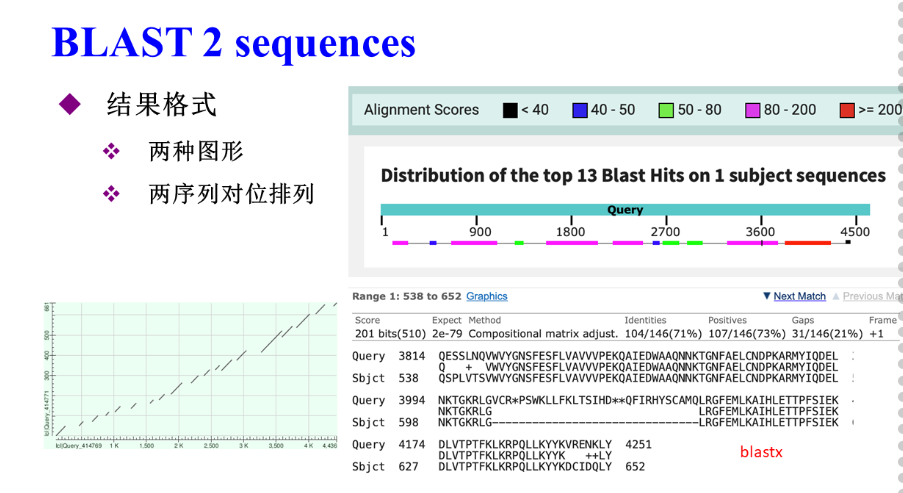
- 分析RNA或DNA的二级结构:(http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi)![[Pasted image 20220529153531.png]]
- BLAST(Basic Local Alignment Search Tool):https://blast.ncbi.nlm.nih.gov/Blast.cgi
- 注意:编码区的比对时,应以密码子为单位,勿改变编码框。
- 基本概念
系统演化分析
- 多序列对位排列(Multiple Sequence Alignment-MSA):让可能多的相似的序列进行比对重复
- 动机:做一致性查询(看是否有保守区域);结构预测(通过一个已知序列的蛋白质预测其他与其保守的蛋白结构);检索蛋白家族的特殊图谱;检索基因的重叠群;进行系统演化分析。
- 目的:
- 功能预测分析:描述一组序列之间的相似性关系;了解一个基因家族的基本特征;寻找motif;保守区域等—用于预测新序列的二级和三级结构,进而推测其生物学功能。
- 系统发育分析:用于描述同源序列之间的亲缘关系的远近,应用到分子进化分析中,是构建分子进化树的基础。
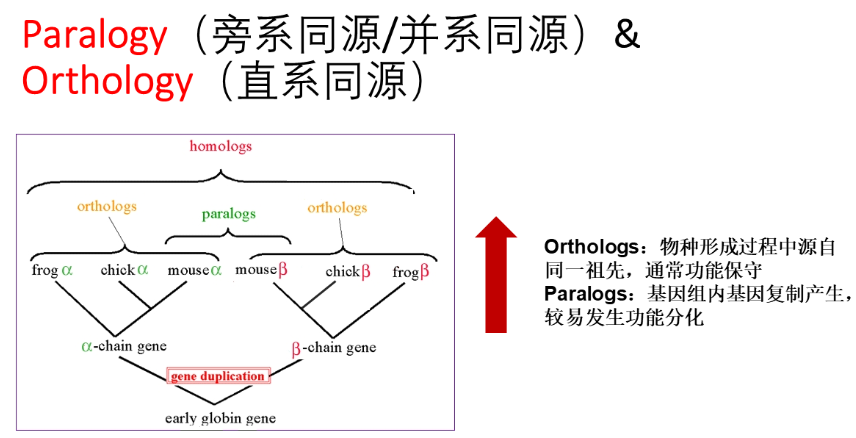
- 变异/共线性分析
- 测序组装分析:
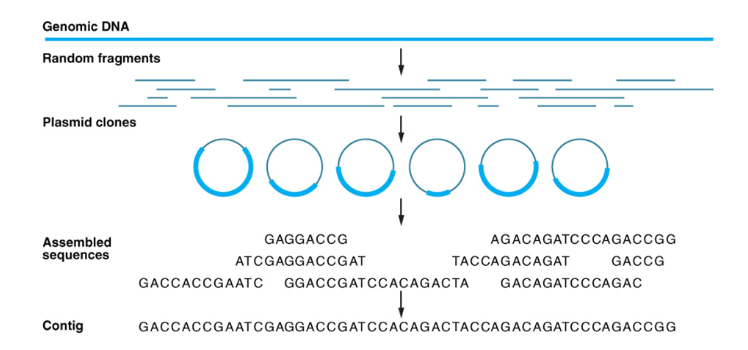
- 怎么做MSA?
- 改进算法(Clustal):http://www.clustal.org/

-
Clustal Omega在线分析:(https://www.ebi.ac.uk/Tools/msa/clustalo/)![[Pasted image 20220530181649.png]]注意:两个星号:代表单一的保守位点;一个冒号代表有比较强的相似性;一个点.代表弱相似性
-
Clustal W/X算法基础:
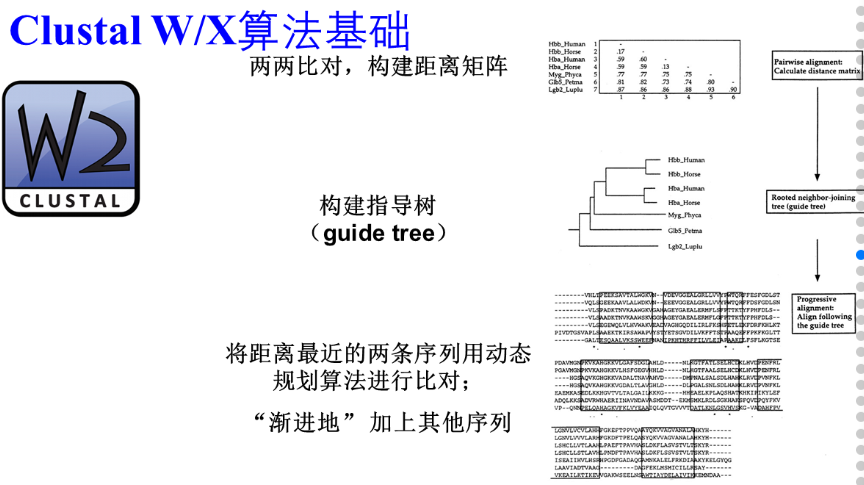
-
- 进一步对排列好的序列进行修饰
1. ESPript—可突出相同或相似的位点(http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi)![[Pasted image 20220530183809.png]]注意:下载没有反应将结果复制到记事本,再改后缀为.aln
2. GeneDoc:https://genedoc.software.informer.com/2.7/sssss

- 改进算法(Clustal):http://www.clustal.org/
- 系统演化分析(Phylogenetic analysis):分析基因或蛋白质的演化(亲缘)关系,系统发生(演化)树
- 系统发生树术语:
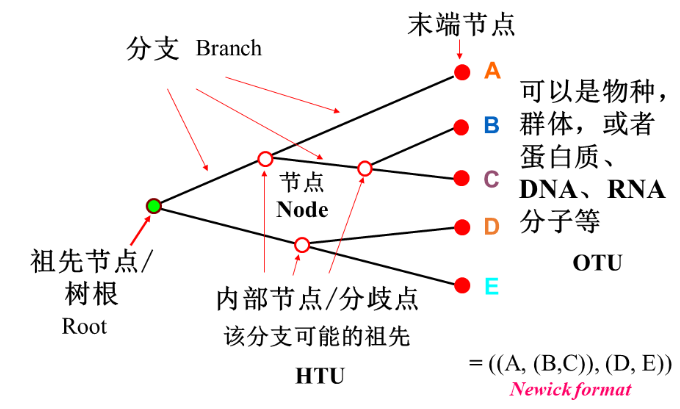
- 演化支:
 注意:演化树树枝长度代表遗传改变信息,超度量树树枝长度代表演化时间
注意:演化树树枝长度代表遗传改变信息,超度量树树枝长度代表演化时间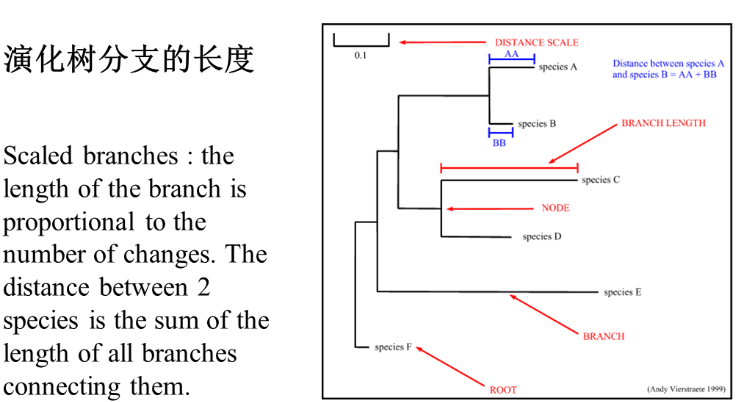
- 有根树和无根树:
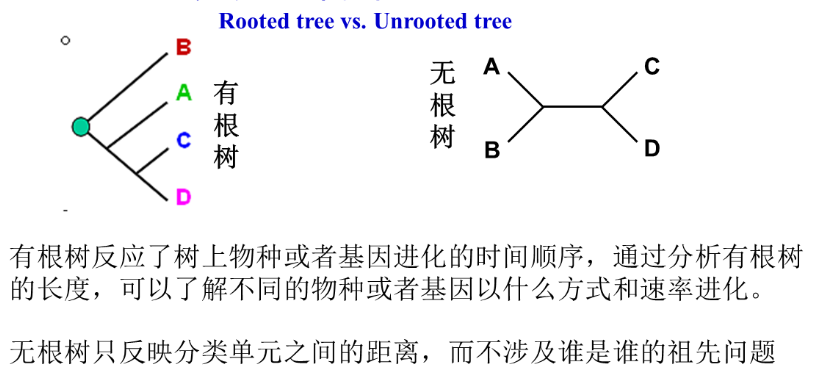
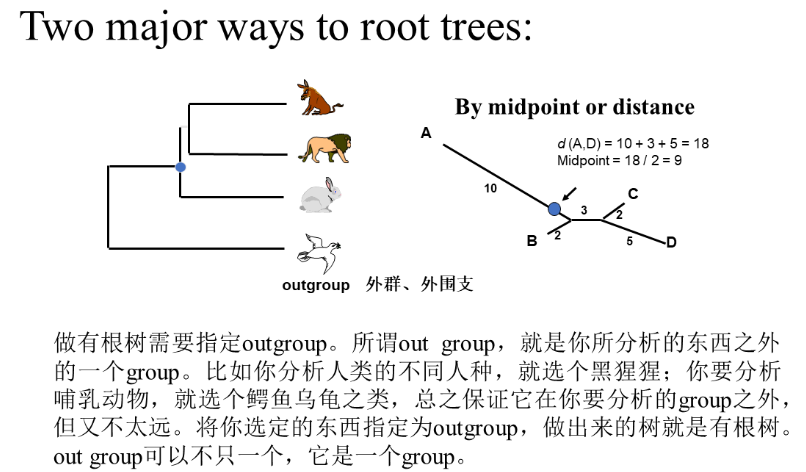
- 演化支:
- 系统发生树术语:
- 系统发育树:
- 定义:系统发育树又称演化树,是表明被认为具有共同祖先的各物种间演化关系的树。在树中每个节点代表其各个分支的最近共同祖先,而节点的线段长度对应了其演化的距离。
- 构建步骤:

- 注意:MSA是构建分子演化树的关键步骤,用于构建演化树的序列必须是同源序列。
- 构建方法:
- 分子演化树构建(ClustalW):https://www.ebi.ac.uk/Tools/phylogeny/simple_phylogeny/
 注意:看图工具可选择NCBI‘s Tree Viewer(https://www.ncbi.nlm.nih.gov/projects/treeview/index.html)
注意:看图工具可选择NCBI‘s Tree Viewer(https://www.ncbi.nlm.nih.gov/projects/treeview/index.html)
- 分子演化树构建(ClustalW):https://www.ebi.ac.uk/Tools/phylogeny/simple_phylogeny/
- 分子演化分析软件MEGA
* 算法比ClustalW多:
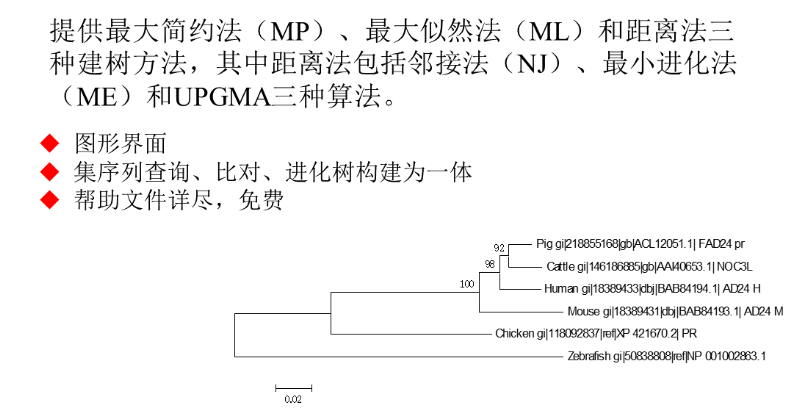 * 可评估进化树的可靠性:自展法
* 可评估进化树的可靠性:自展法
基因分析和基因组注释
- 基因结构:
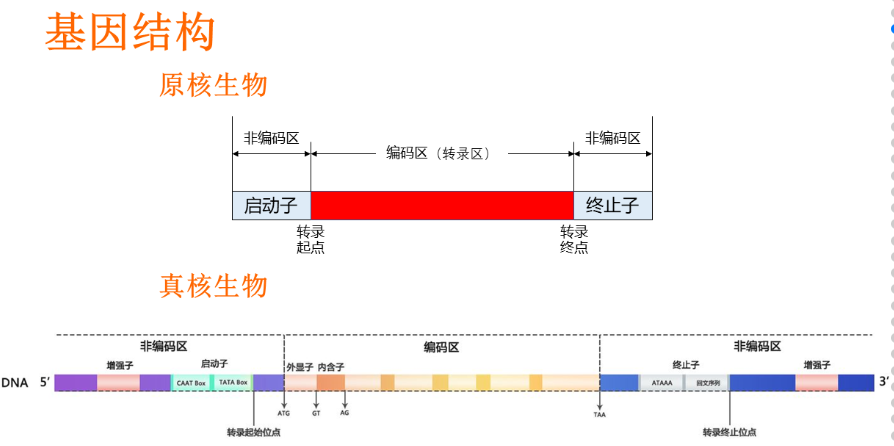
- 基因预测和基因结构分析的作用:预测编码蛋白质的基因(Protein-coding gene);预测非编码RNA基因(Non-coding RNA gene)
- 基因预测的基本分析内容:排除重复序列;确定基因结构(开放阅读框、基因的调控区-启动子)
- *已知mRNA开放读码框(ORF)的确定:(https://www.ncbi.nlm.nih.gov/orffinder/)![[Pasted image 20220530204232.png]]注意:只适合于原核生物
- 基因预测的基本方法:
- 序列相似性搜索:

- 模式序列预测基因:基于一个或多个已知序列模式对未知序列进行分类(启动子结构;外显子、内含子),密码子偏爱性,基因结构预测等(多种物种的参照)
- Gene Finding:(http://www.softberry.com/ )

- GENSCAN:(http://hollywood.mit.edu/GENSCAN.html)![[Pasted image 20220530210357.png]]
- GeneMark:用于真核、原核和病毒等基因的预测(http://exon.gatech.edu/GeneMark/)
- Augustus:用于真核基因的预测,可做全基因组基因预测和功能注释,可预测可变剪切(http://bioinf.uni-greifswald.de/augustus/)![[Pasted image 20220530211120.png]]
- 利用比较基因组预测基因:(http://mblab.wustl.edu/nscan/)![[Pasted image 20220530211500.png]]
- 常见问题:

- 序列相似性搜索:
- 基因精细结构分析:
- NNPP分析转录起始位点:(http://www.fruitfly.org/seq_tools/promoter.html)
- 启动子预测:Promoter 2.0 Prediction Server(https://services.healthtech.dtu.dk/service.php?Promoter-2.0)![[Pasted image 20220530212536.png]]
- 分析转录因子结合位点:
- JASPAR:(http://jaspar.genereg.net/)![[Pasted image 20220530212917.png]]
- PlantPAN:(http://plantpan.itps.ncku.edu.tw/index.html)
- 其他软件:
- 非编码RNA基因的预测:非编码RNA(Non-coding RNA)是指转录但不编码蛋白质的RNA,包括rRNA、tRNA、snRNA、snoRNA、microRNA等多种已知功能的RNA。
 工具:
工具:
- miRNA:miRBase:(http://www.mirbase.org/)—可利用miRNA编号或者关键词检索;利用物种染色体位置检索。检索内容为***miRNA家族,已知序列预测是否含有miRNA***
- MicroRNA靶基因的预测:

- ENCODE:DNA元件的百科全书
 基因预测或基因结构分析原则:
基因预测或基因结构分析原则:
- Smilarity-based or Comparative:BLAST—判断其他物种是否有相似的序列?
- Ab initio=“From the beginning”
- Combined “evidence-based”—综合多种分析方法
蛋白质分析
- 分析意义:
- 序列相似的蛋白质具有相似的三维结构—可预测结构
- 不同蛋白质中相同的结构域(domain)具有相似的功能—预测功能
- 分析蛋白质的功能
- 一级结构:氨基酸的排列顺序
- 二级结构:主要由氢键维系的结果(α-helix、β-sheet)
- 三级结构:二级结构进一步折叠形成的结构域
- 分析工具:
- ExPASy(Expert Protein Analysis System):(https://www.expasy.org/)![[Pasted image 20220531163734.png]]
- CBS(Center for Biological Sequence Analysis):(http://www.cbs.dtu.dk/services/)![[Pasted image 20220531164112.png]]新地址:(https://services.healthtech.dtu.dk)
- 蛋白质性质和结构分析其他软件:

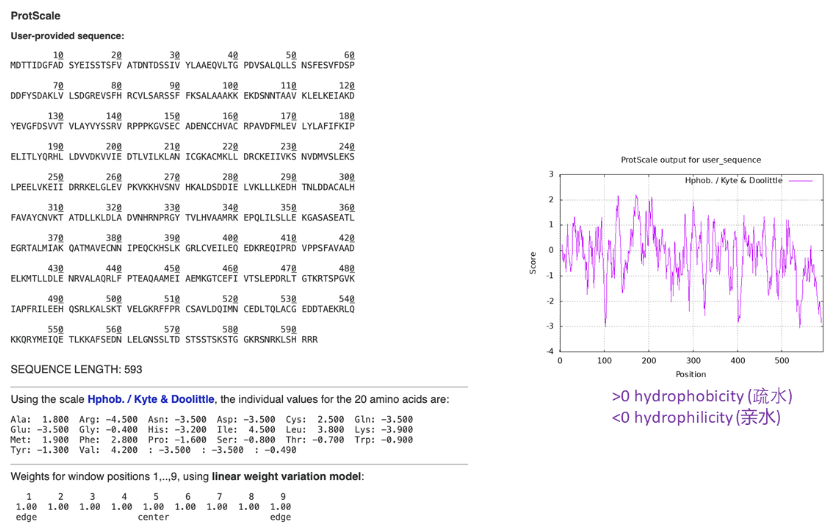
- 分析蛋白质的一级结构:分析蛋白质的氨基酸组成、等电点(pI)、分子量(molecular weight, Mw)、疏水性(hydrophobicity)等
- 分析蛋白质的pI、Mw、氨基酸组成:(https://web.expasy.org/protparam/)![[Pasted image 20220531170939.png]]
- 分析蛋白质的疏水性:(https://web.expasy.org/protscale/)![[Pasted image 20220531171217.png]]结果分析:
- 分析蛋白质的模体(MOTIFS):ScanProsite(https://prosite.expasy.org/scanprosite/)![[Pasted image 20220531171812.png]]
- 分析蛋白质的保守结构域:(https://prosite.expasy.org/)![[Pasted image 20220531172040.png]]
- 其他预测蛋白质结构域的工具:

- 分析蛋白的二级结构:—α-helix、β-sheet、turn loop、random coil
- SOPMA:(https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html)![[Pasted image 20220531192521.png]]
- 分析蛋白质的三级结构
- 根据已知蛋白质结构推测未知蛋白质结构:
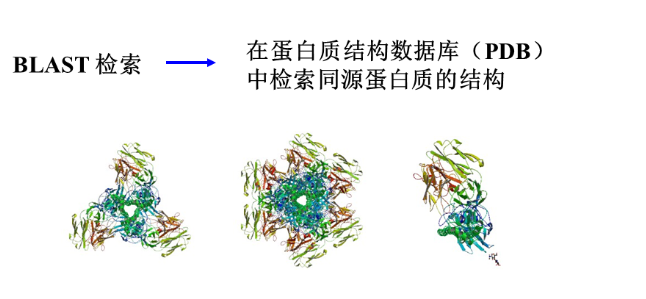
- 通过分子建模(molecular modeling)分析蛋白质结构— 分析复杂、适用于专业人员
- Phyre2:直接输入蛋白质序列和E-mail,2小时后反馈结果,同时给出与之同源的蛋白质的3D结构(http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index)
- SWISS-MODEL Workspace:提供多种分子建模方式(https://swissmodel.expasy.org/interactive)
- AlphaFold Protein Structure Database:

- 注意:结构预测流程:
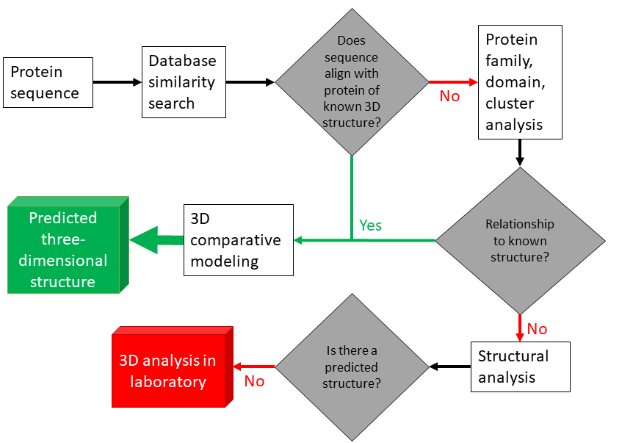
- 根据已知蛋白质结构推测未知蛋白质结构:
- 分析膜蛋白质:
- 预测膜整合蛋白的跨膜区:SOSUI(http://harrier.nagahama-i-bio.ac.jp/sosui/)![[Pasted image 20220531195007.png]]
- 其他跨膜分析软件:

- 分析膜锚定蛋白的GPI位点:(https://mendel.imp.ac.at/))![[Pasted image 20220531195458.png]]
- For Plant Proteins:http://mendel.imp.ac.at/gpi/plant_server.html
- For Animals Proteins:http://mendel.imp.ac.at/gpi/gpi_server.html
- For Fungal Proteins: http://mendel.imp.ac.at/gpi/fungi_server.html
- 分析蛋白质的翻译后修饰
- 分析信号肽及其剪切位点:DTU HEALTH TECH(https://services.healthtech.dtu.dk/service.php?SignalP)![[Pasted image 20220531203739.png]]其他网站:

- 分析糖链连接点
- O-连接糖蛋白:(https://services.healthtech.dtu.dk/service.php?NetOGlyc-4.0)![[Pasted image 20220531210705.png]]
- N-连接糖蛋白:(https://services.healthtech.dtu.dk)![[Pasted image 20220531210903.png]]
- 分析蛋白质修饰位点:

- 分析信号肽及其剪切位点:DTU HEALTH TECH(https://services.healthtech.dtu.dk/service.php?SignalP)![[Pasted image 20220531203739.png]]其他网站:
- 分析蛋白质的亚细胞定位(Uniport):—有助于了解蛋白质功能与蛋白质互作
- PSORT:(https://wolfpsort.hgc.jp/))![[Pasted image 20220531212312.png]]
- 其他:
- 分析化学因子作用蛋白质的位点:PeptideCutter(https://web.expasy.org/peptide_cutter/)![[Pasted image 20220531212516.png]]
基因组浏览器
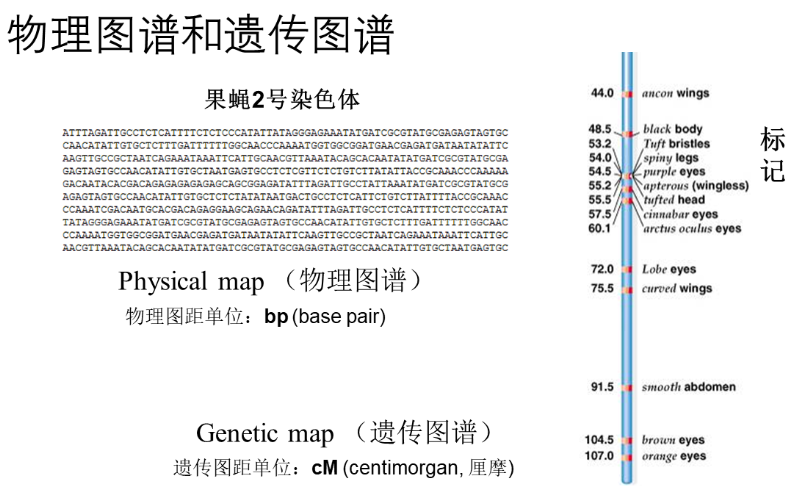
- 关于物理图谱和遗传图谱:
- 基因组浏览器:
- UCSC基因浏览器:(http://genome.ucsc.edu/)![[Pasted image 20220601092920.png]]

- Ensembl基因组浏览器:(https://asia.ensembl.org/index.html)![[Pasted image 20220601093249.png]]
- 代表性模式生物浏览器:
- MGI(Mouse Genome Informatics):(http://www.informatics.jax.org/)![[Pasted image 20220601093514.png]]
- Flybase:(https://flybase.osrg/)![[Pasted image 20220601093750.png]]
- SGD(Saccharomyces Genome Database):酿酒酵母数据库(http://yeastgenome.org/)—包括基因组注释、表达谱、蛋白互作、文献、分析工具
- tair(The Arabidopsis Information Resource):(https://www.arabidopsis.org/)![[Pasted image 20220601103944.png]]
- 水稻(rap-db):(https://rapdb.dna.affrc.go.jp)![[Pasted image 20220601104148.png]]
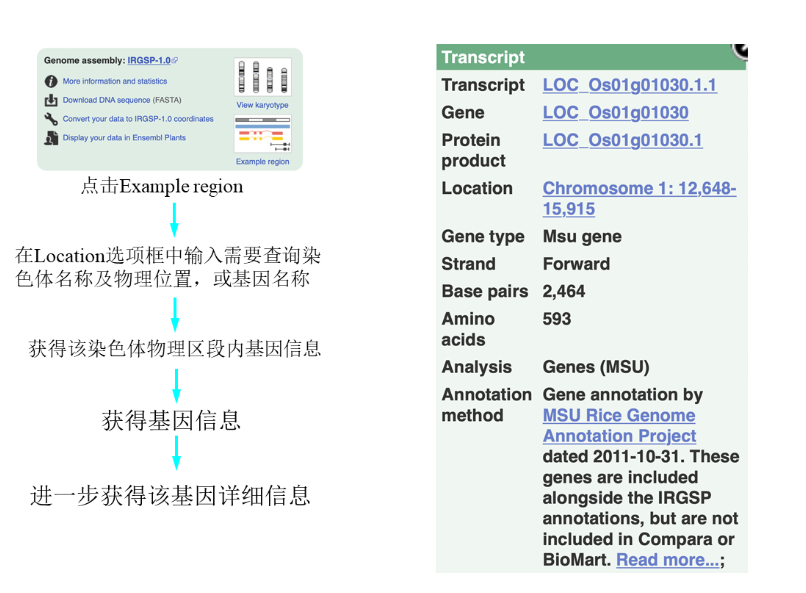
- 综合植物模式生物数据库(ensembl):(https://ensembl.gramene.org/index.html)![[Pasted image 20220601104359.png]]例如:


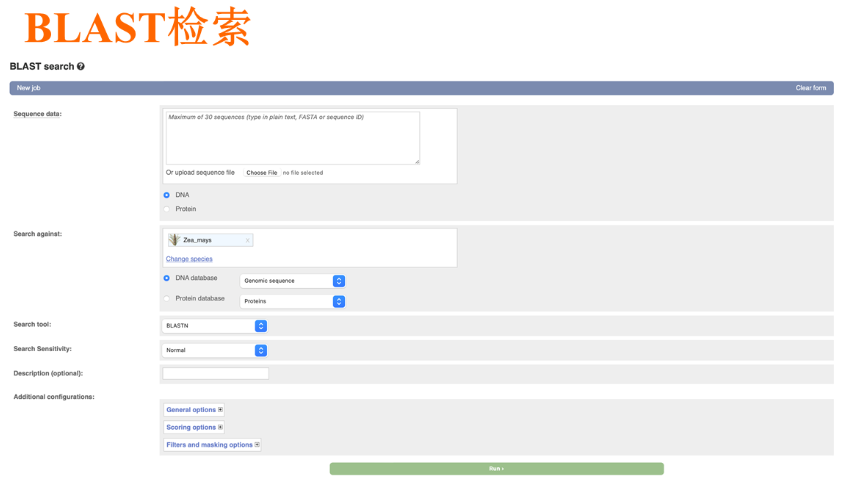
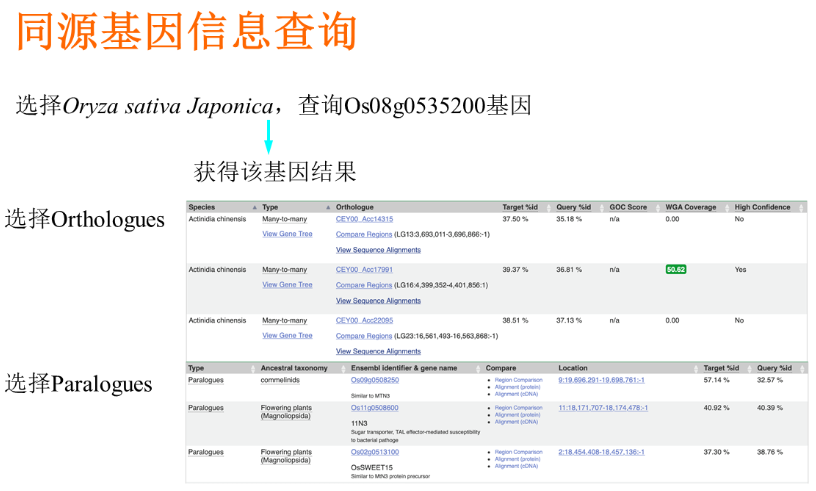
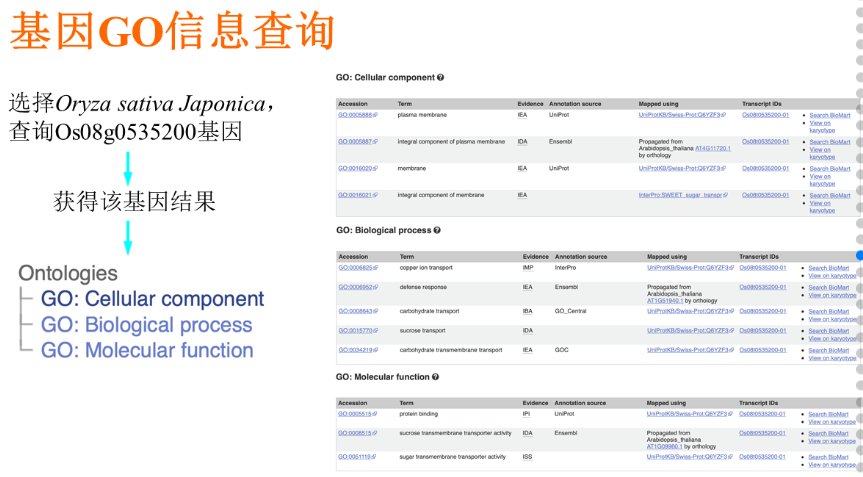
- 转录组检索:(https://www.ebi.ac.uk/gxa/home#)![[Pasted image 20220601105833.png]]
- 其他重要农作物数据库:


- 代谢路径浏览器:http://pathway.gramene.org/

- 家畜、家禽基因组浏览器:(https://www.animalgenome.org/bioinfo/)![[Pasted image 20220601111051.png]]
- UCSC基因浏览器:(http://genome.ucsc.edu/)![[Pasted image 20220601092920.png]]
生物信息学其他应用
- 芯片表达谱及RNA-seq应用:
-
芯片定义:固定着大规模的DNA片段,用于检测整个转录组内成千上万个基因的mRNA表达水平变化的微型器材
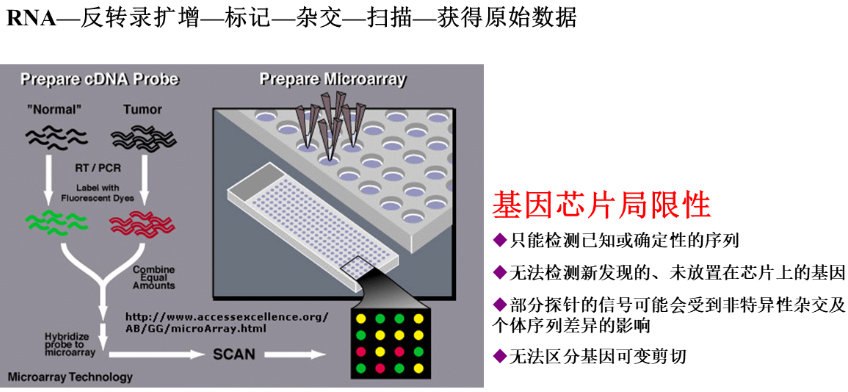
-
RNA-sequencing:基于测序技术的转录组学研究方法,把mRNA、smallRNA、non-coding RNA等或者其中一些用高通量测序技术把它们的序列测出来,反应它们的表达水平*。



-
ChIP-seq应用:ChIP-chromatin immunoprecipitation with massively parallel DNA sequencing(染色质免疫共沉淀测序),利于分析鉴定转录因子靶位点,以及基因组上组蛋白及其各种修饰


-
Galaxy应用:各种方法获得核酸样本建库,并深度测序(https://usegalaxy.org/)![[Pasted image 20220601141007.png]]
-
GEO数据库应用:(https://www.ncbi.nlm.nih.gov/geo/)![[Pasted image 20220601141231.png]]
 注意:利用GEO Profile进行关键词检索。
注意:利用GEO Profile进行关键词检索。 -
常用的综合表达谱数据库:GEO、Expression Atlas(http://www.ebi.ac.uk/gxa/)—可直接通过探针编号、基因名称等关键词查询,也可以通过序列获得探针编号
-
- CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats) gRNA设计:
 例如:tefor(http://crispor.tefor.net/)
例如:tefor(http://crispor.tefor.net/) - Primer设计:
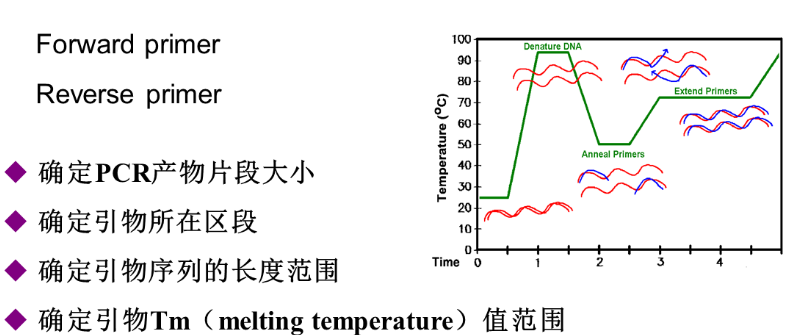 例如:*Primer3web version 4.1.0:https://bioinfo.ut.ee/primer3/
例如:*Primer3web version 4.1.0:https://bioinfo.ut.ee/primer3/ 
- 限制性核酸内切酶切割位点分析:
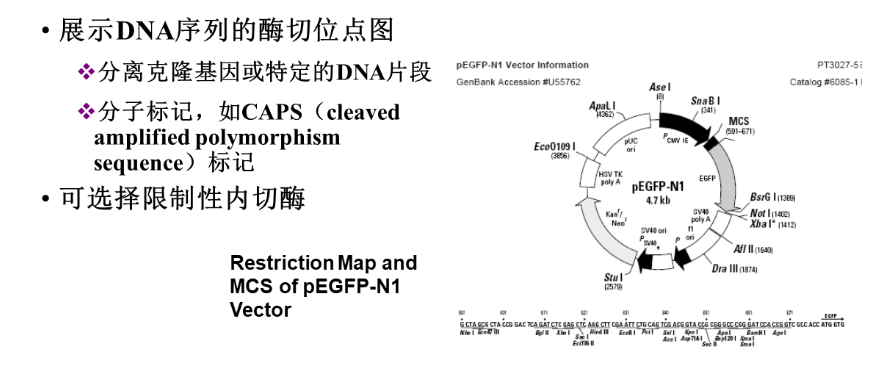
- NEBcutter:(http://nc2.neb.com/NEBcutter2/)![[Pasted image 20220601194035.png]]
- RestrictionMapper:(http://www.restrictionmapper.org)![[Pasted image 20220601194158.png]]
- 测序在组学中的应用: